The Eraser Hypothesis: Epitranscriptomic Modulation of Synaptic Plasticity Under Metabolic Stress
Joshua Kyan Aalampour
Paris, France
April 13, 2026
Abstract
Nearly every major pharmacologic approach to cognitive enhancement, from racetams to ampakines to BDNF agonists, operates at the neurotransmission level. Each aims to make neurons signal harder. This paper argues that the constraint lies deeper. Memory consolidation requires local protein synthesis at activated synapses, and the translation of plasticity-encoding messenger RNAs is gated by a chemical mark called N6-methyladenosine (m6A), installed by writer enzymes and removed by erasers. The eraser FTO strips m6A from plasticity transcripts, suppressing their translation. Under chronic metabolic stress (obesity, insulin resistance), FTO-mediated erasure appears to increase and total brain m6A levels decline, tightening a brake that should respond to context rather than run constantly. Yet complete FTO loss damages neurogenesis and impairs certain memory processes. We propose that partial, allosteric modulation of FTO through a recently identified cryptic site at the domain interface, rather than the conserved catalytic center, could partially restore plasticity-gene translation under metabolic stress without the liabilities of broad enzymatic shutdown.
1. The Volume Knob Problem
For half a century, the search for cognitive enhancers has been a search for louder signals. Piracetam modulates AMPA receptors. Modafinil acts on dopamine and orexin pathways. Ampakines potentiate glutamate signaling. BDNF agonists activate TrkB.
The pharmacology varies, but the logic does not. Nearly every intervention operates at the level of neurotransmission, and none has reliably delivered lasting improvement in learning or memory in the populations where cognitive decline is most pressing. The ceiling is conceptual.
Memory formation requires more than electrical signaling. When a synapse strengthens during long-term potentiation (LTP), new proteins must be built locally at the activated synapse to stabilize the change (Sutton and Schuman, 2006). The messenger RNAs for these proteins (Arc, CaMKII alpha, BDNF among them) sit at synapses already and are translationally silent. Learning triggers their translation. Without local protein synthesis, long-term memory does not form.
Translation-level interventions exist: ISRIB and related integrated-stress-response modulators enhance memory by relieving global translational repression (Sidrauski et al., 2013). But global relief is not selective control. No existing approach targets the transcript-specific, epitranscriptomic gating of plasticity-gene translation. Picture a construction site where workers, blueprints and materials are all present, but a regulatory checkpoint keeps rejecting the permits. The constraint is the checkpoint.
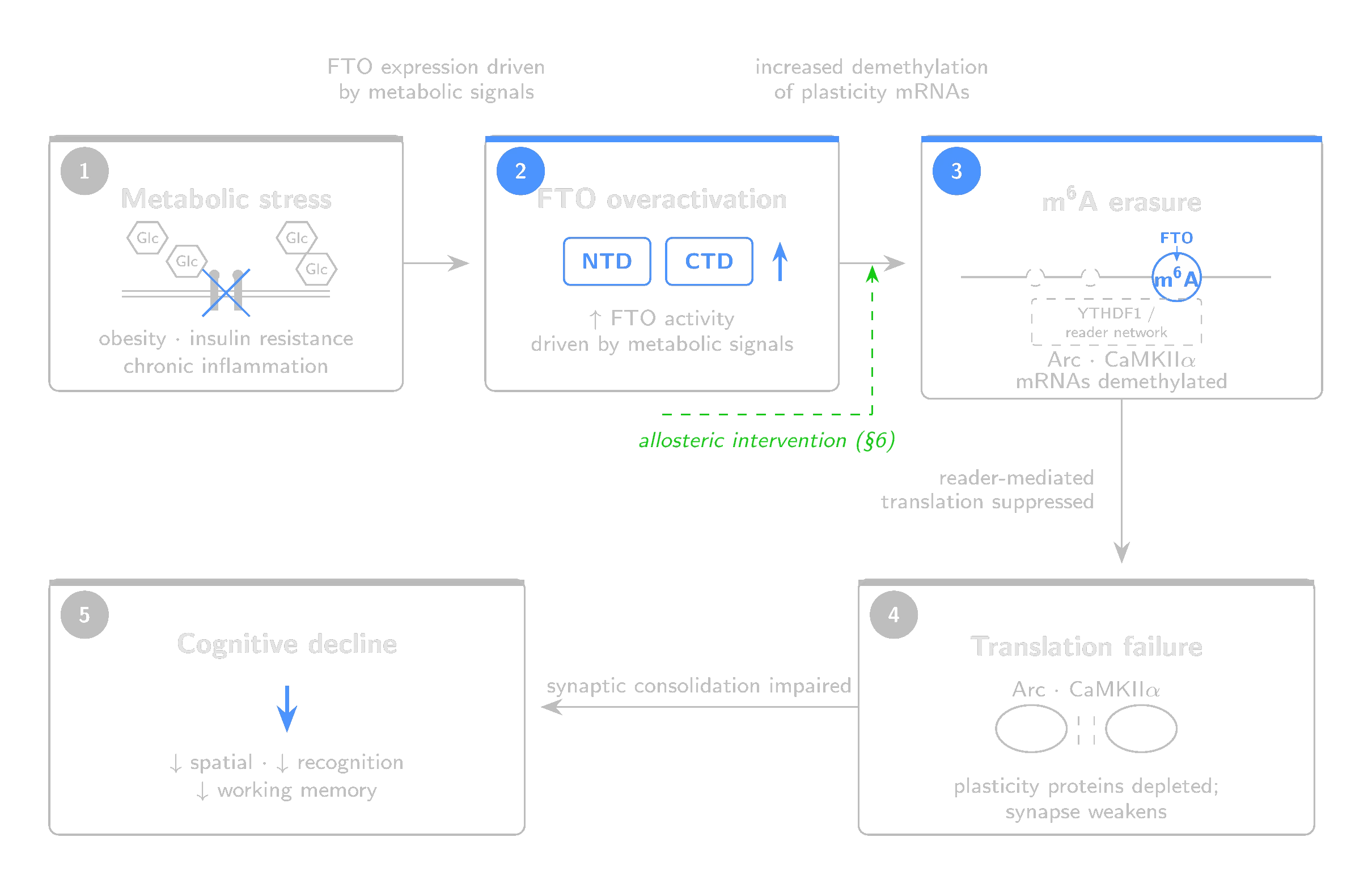
In the brain, the permits are chemical marks on messenger RNA. The checkpoint is the epitranscriptome. The enzyme responsible for removing those marks, FTO, becomes overactive under the same conditions (obesity, insulin resistance, chronic inflammation) where cognitive decline is most prevalent. This paper argues that the next intervention point is at the level of RNA, through an epitranscriptomic mechanism no established cognitive-enhancer class has exploited.
2. The Epitranscriptomic Layer
The cell's real-time regulatory decisions happen on messenger RNA, through chemical modifications that determine whether a transcript is translated, stored or destroyed. The most common of these is N6-methyladenosine (m6A): a methyl group on the nitrogen at the sixth position of adenosine. It is the most abundant internal modification on eukaryotic mRNA (Dominissini et al., 2012), and it is reversible (Zheng et al., 2013; Jia et al., 2011). DNA methylation persists across cell divisions. m6A marks are written and erased in minutes to hours. In signal processing terms, DNA methylation is firmware while m6A is RAM.
The system has three classes of proteins. Writers (the METTL3/METTL14 complex) install m6A on target transcripts. Readers interpret the mark: YTHDF1 binds m6A-marked mRNAs and drives their translation (Wang et al., 2015). For plasticity-related mRNAs, YTHDF1-mediated translation is a key pathway in hippocampal learning (Shi et al., 2018). Erasers remove the mark. FTO, an Fe(II)/2-oxoglutarate-dependent dioxygenase, oxidizes the methyl group off adenosine through a stepwise reaction:
m6A --[FTO, O2, 2-oxoglutarate]--> hm6A --> f6A --> A
The methyl group passes through hydroxymethyl (hm6A) and formyl (f6A) intermediates before full removal. The chemistry requires molecular oxygen and 2-oxoglutarate, producing succinate and CO2. ALKBH5, the only other known mammalian m6A eraser, removes the methyl group directly without stable oxidized intermediates (Zheng et al., 2013).
Three genetic results connect m6A to memory. METTL3 (the writer) knockout in the adult mouse hippocampus impairs long-term memory consolidation while leaving short-term memory intact (Zhang et al., 2018). m6A levels on plasticity-related transcripts increase after learning (Widagdo et al., 2016). YTHDF1 (the reader) knockout impairs hippocampus-dependent learning and memory (Shi et al., 2018). The causal chain: writer installs mark, reader drives translation, plasticity proteins are built, memory consolidates.
FTO also demethylates cap-adjacent m6Am (Mauer et al., 2017), complicating substrate attribution. This ambiguity is revisited in Section 4.
3. FTO in the Brain
FTO (fat mass and obesity-associated protein) is an RNA demethylase (Jia et al., 2011), one of only two known m6A erasers in mammals (Jia et al., 2011; Zheng et al., 2013).
FTO is expressed broadly in the brain, including hippocampus and prefrontal cortex (Widagdo et al., 2016; Walters et al., 2017), regions central to memory formation. Plasticity-related transcripts show learning-linked m6A dynamics consistent with FTO involvement (Widagdo et al., 2016), though direct substrate assignment at the individual-transcript level is less established. FTO also demethylates cap-adjacent m6Am (Section 2), and which substrate class predominates may depend on subcellular localization; the mechanism proposed here assumes internal m6A on plasticity transcripts is the operative target. This assumption remains to be tested directly. By stripping their m6A marks, FTO reduces YTHDF1-mediated translation. Less m6A means less reader binding, which results in less protein at the synapse.
In a healthy brain, FTO is part of the write-erase cycle that gives m6A its temporal precision. Neural activity shifts the balance of writing and erasing, producing transient pulses of enhanced translation that return to baseline. Without the eraser, marks would accumulate and translation would become constitutive rather than activity-dependent, which defeats the purpose of local protein synthesis. FTO expression is itself activity-regulated (Walters et al., 2017), so the system is self-correcting under normal conditions.
The problem arises when metabolic signals, rather than learning, may drive FTO up.
4. The Metabolic Context
Metabolic syndrome is associated with progressive cognitive decline not fully explained by classical Alzheimer's disease pathology. The decline is subtler than dementia. What you get is a gradual erosion of learning efficiency, working memory and executive function that begins years before clinical diagnosis. What has been missing is a molecular mechanism connecting the metabolic insult to the plasticity deficit. FTO may provide one.
In HFD-fed SAMP8 mice, FTO inhibition with FB23 increased total brain m6A levels, upregulated plasticity-related genes (Arc, cFos, Bdnf, Ngf) and improved structural measures including neurite length and spine density (Irisarri et al., 2025). Cognitive performance on spatial memory tasks recovered. The rescue was not confined to the brain, however: FB23 also corrected peripheral metabolic markers, leaving open whether the cognitive benefit is central, peripheral, or both. In the same study, Ythdf1/2/3 expression decreased while Ythdc1/2 expression increased after FB23 treatment. This complicates the linear model in which YTHDF1 is the sole translational effector of m6A marks on plasticity transcripts. It also suggests that the cognitive rescue may involve YTHDC-mediated nuclear processing, alternative reader recruitment, or compensatory shifts in translational efficiency rather than a simple restoration of YTHDF1-driven translation. The plasticity readouts in this study were drawn from cortical tissue, not direct measurements of hippocampal FTO activity. Direct causal evidence linking FTO modulation to cognitive rescue under metabolic stress currently depends on a small number of model systems, the SAMP8-HFD study chief among them.
FTO's second substrate class, cap-adjacent m6Am, adds a layer of unresolved complexity. Under high-fat diet and obesity conditions, m6Am levels tracked metabolic regulation more closely than internal m6A in at least one study (Ben-Haim et al., 2021). This raises the possibility that some of FTO's metabolic-cognitive effects operate through mRNA stability and translation-initiation pathways rather than through internal m6A alone. Whether the cognitive rescue observed in the SAMP8-HFD model was driven primarily by restored internal m6A, restored m6Am, or both has not been resolved. Disentangling these substrate contributions is an open experimental question.
5. The Contradictions
The argument so far runs clean: metabolic stress pushes FTO up, cognition comes down, FTO inhibition reverses the decline. However, the broader literature is less cooperative.
Knocking down FTO in the medial prefrontal cortex enhanced fear-memory consolidation (Widagdo et al., 2016). Reducing FTO in the dorsal hippocampus also enhanced contextual fear memory (Walters et al., 2017). Yet FTO deficiency in mice impairs working memory and disrupts BDNF processing (Spychala and Ruther, 2019), and genetic knockout in adult neural stem cells damages neurogenesis (Li et al., 2017). Pharmacologic FTO inhibition with meclofenamic acid in the hippocampus disrupts novel-object-recognition reconsolidation via the BDNF-TrkB pathway (Chang et al., 2023). Yet in the SAMP8-HFD model, FTO inhibition rescued cognition (Irisarri et al., 2025).
Even within the hippocampus, outcomes diverge depending on the paradigm, the method of manipulation and the metabolic context. The outcomes track the method, the region and the starting state.
5.1 Four Distinctions
The conflict dissolves once you stop treating "FTO inhibition" as a single intervention.
Genetic ablation is not pharmacological modulation. Knocking out a gene removes the protein entirely, permanently and from every subcellular compartment. A drug that partially inhibits an enzyme for a few hours is a fundamentally different perturbation. The neurogenesis deficit in FTO knockout mice (Li et al., 2017) tells us that stem cells need some FTO to function. It does not tell us that, say, a 40 percent reduction in catalytic activity for six hours would produce the same effect. Genetic studies only define the boundaries of what FTO does. They do not predict the pharmacological window.
Complete loss is not partial reduction. Even among pharmacological studies, saturating an enzyme's active site is different from partially shifting its conformational equilibrium. The studies showing harm used either genetic tools or competitive inhibitors. Whether partial inhibition preserves the beneficial functions while reducing the pathological excess has not been tested, because the tools to do it have not existed.
The healthy brain is not the metabolically stressed brain. In a healthy animal, FTO is at homeostatic levels. Inhibiting it disrupts a system that is working correctly. In a metabolically stressed animal, FTO is pathologically overactive. Inhibiting it corrects an excess. The same intervention applied to two different starting states produces opposite outcomes.
Brain region and paradigm matter. Reducing hippocampal FTO can enhance contextual fear memory (Walters et al., 2017) while disrupting object-recognition reconsolidation (Chang et al., 2023) and, in genetic models, impairing working memory (Spychala and Ruther, 2019). Whether the net effect of systemic modulation is beneficial depends on which circuit is most dysregulated and which cognitive domain is being assessed.
5.2 The Refined Hypothesis
The hypothesis is narrower than "FTO inhibition improves cognition," which is a claim the data do not support.
Partial, context-biased modulation of FTO, reducing pathological overactivity without eliminating basal function, can separate the metabolic-stress cognitive benefit from the broad-loss liabilities observed in genetic and high-occupancy pharmacological studies.
Three testable predictions follow:
1. A partial FTO modulator should rescue cognitive deficits in metabolically stressed animals while producing minimal effects in healthy controls.
2. The therapeutic window should be wider than that of competitive active-site inhibitors, because partial modulation has a built-in ceiling.
3. The compound should preserve neurogenesis and hippocampal function at doses that correct the metabolic-stress phenotype, because basal FTO activity is maintained.
Testing these predictions requires a pharmacological tool that has not previously been available. Until recently, every reported FTO inhibitor was a competitive active-site binder producing dose-dependent, binary inhibition.
6. The Allosteric Proposition
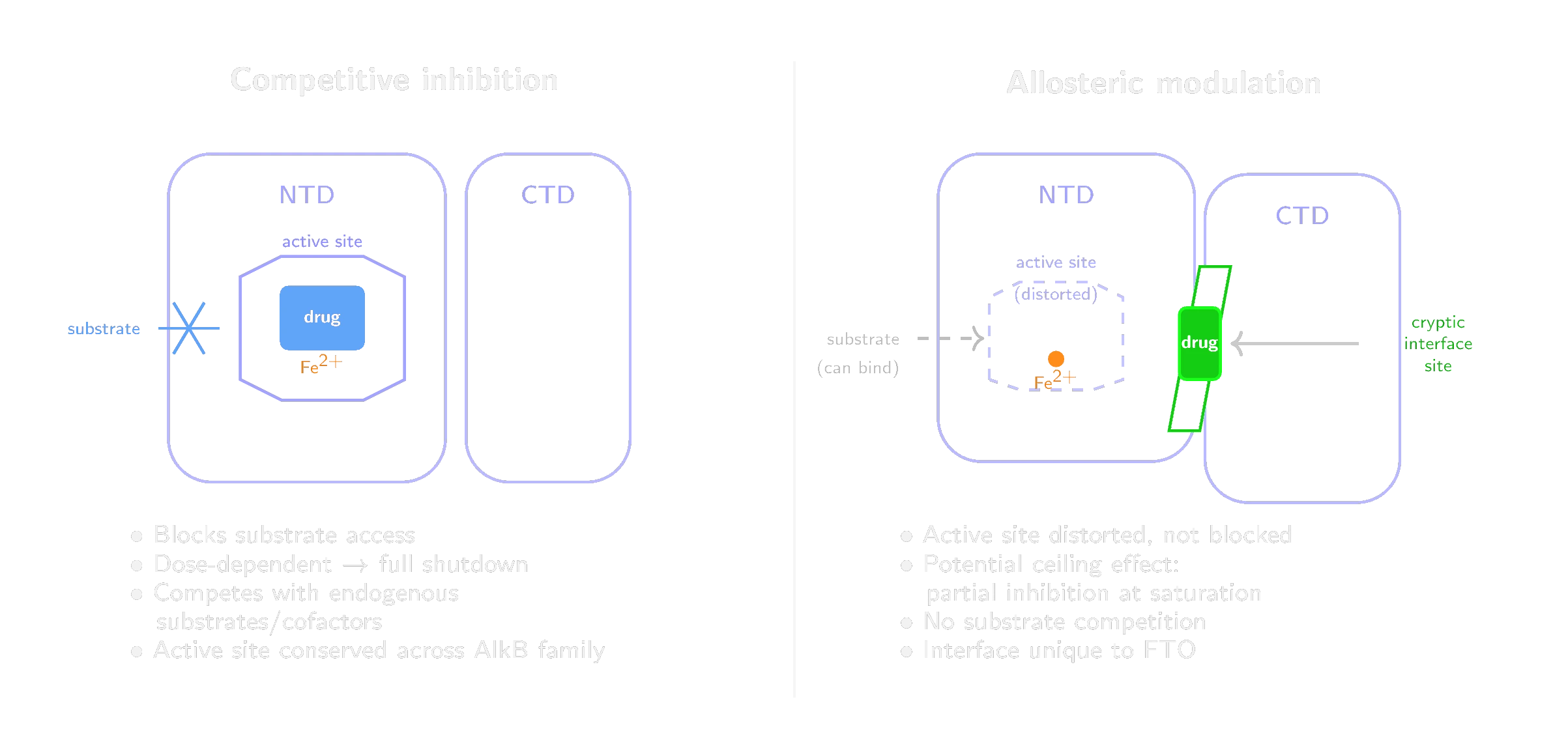
Most previously reported FTO inhibitors enter the active site, compete with the natural substrates and block catalysis. This is competitive inhibition. It creates three problems, each with a corresponding answer at a different site on the protein.
Selectivity. The FTO active site belongs to the AlkB dioxygenase superfamily. Its iron-binding and 2-oxoglutarate-binding residues are conserved within the AlkB subfamily (FTO, ALKBH1 through ALKBH8) and, more broadly, across structurally related Fe(II)/2-oxoglutarate oxygenases. FB23-2 was recently found to also inhibit DHODH, an entirely different enzyme class, likely due to structural similarities between the catalytic pockets (Tarullo et al., 2024). The NTD-CTD domain interface is structurally specific to FTO. ALKBH5 has a different domain organization. Other AlkB members lack this interface entirely. A molecule designed for it has no analogous site to occupy on the off-targets that derailed FB23-2.
Dose-response. Competitive inhibitors produce a simple relationship between concentration and inhibition: as concentration rises, activity approaches zero. For FTO, where complete shutdown damages neurogenesis and impairs hippocampal function (Section 5), this is the wrong profile. The therapeutic index may be narrow. An allosteric modulator that shifts the conformational equilibrium between active and inactive states can produce a ceiling effect, depending on the residual catalytic competence of the bound conformation. If the trapped state retains partial activity, even saturating concentrations slow the enzyme rather than shut it down. The degree of inhibition can be tuned by medicinal chemistry.
Substrate competition. Active-site inhibitors must contend with the endogenous ligand environment at the catalytic center: 2-oxoglutarate is present at roughly 1 mM intracellularly, and the RNA substrate pool is abundant. Achieving sufficient occupancy in a tissue behind the blood-brain barrier compounds the challenge. The allosteric site is physically separate from the active site. Because it binds outside the catalytic center, it need not directly compete with co-substrate or RNA substrate concentrations.
The structural basis for this approach was laid in 2022, when Khatiwada and colleagues used solution NMR and accelerated molecular dynamics to generate a conformational ensemble of apo FTO. This revealed that the domain interface harbors transient druggable pockets not visible in static crystal structures (Khatiwada et al., 2022). In 2026, Singh and colleagues built on this work and reported the first noncompetitive FTO inhibitor targeting one of these pockets (Singh et al., 2026). Using molecular dynamics, solution NMR and kinetic analysis, they characterized a binding site at the NTD-CTD interface that can be stabilized by a small molecule, trapping FTO in a catalytically impaired conformation. The proof-of-concept compound showed noncompetitive kinetics: it reduced FTO's maximal catalytic rate (V_max) without changing the enzyme's affinity for its substrates (K_m). Whether compounds targeting this site cross the blood-brain barrier and preserve neurogenesis at effective doses remains to be shown.
We propose that allosteric modulation of FTO through the cryptic domain-interface site is the pharmacological strategy best matched to the biological problem described in this paper. The liabilities of competitive FTO inhibition (poor selectivity, binary dose-response, narrow therapeutic index) correspond to the advantages of this site (intrinsic selectivity, graded modulation, no substrate competition).
7. Conclusion
Cognitive enhancement has been framed as a neurotransmission problem for fifty years. This paper argues that the constraint is translational. The brain's ability to convert experience into lasting circuit changes depends on local protein synthesis at activated synapses, and that synthesis is gated by m6A marks that are written and erased in response to neural activity. When the eraser FTO is driven into overactivity by chronic metabolic stress, the gate closes too tightly and plasticity suffers. Allosteric modulation through the recently discovered cryptic domain-interface site offers a pharmacological strategy matched to the biology. The approach is partial, selective, graded, and structurally free of the active-site liabilities that have limited every competitive FTO inhibitor to date. The hypothesis is specific and falsifiable. What remains is the chemistry.
References
[1] M. A. Sutton and E. M. Schuman. Dendritic protein synthesis, synaptic plasticity, and memory. Cell, 127:49-58, 2006.
[2] C. Sidrauski, D. Acosta-Alvear, A. Khoutorsky, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife, 2:e00498, 2013.
[3] D. Dominissini, S. Moshitch-Moshkovitz, S. Schwartz, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature, 485:201-206, 2012.
[4] G. Zheng, J. A. Dahl, Y. Niu, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Molecular Cell, 49:18-29, 2013.
[5] G. Jia, Y. Fu, X. Zhao, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nature Chemical Biology, 7:885-887, 2011.
[6] X. Wang, B. S. Zhao, I. A. Roundtree, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell, 161:1388-1399, 2015.
[7] H. Shi, X. Zhang, Y.-L. Weng, et al. m6A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature, 563:249-253, 2018.
[8] Z. Zhang, M. Wang, D. Xie, et al. METTL3-mediated N6-methyladenosine mRNA modification enhances long-term memory consolidation. Cell Research, 28:1050-1061, 2018.
[9] J. Widagdo, Q.-Y. Zhao, M.-J. Kempen, et al. Experience-dependent accumulation of N6-methyladenosine in the prefrontal cortex is associated with memory processes in mice. Journal of Neuroscience, 36:6771-6777, 2016.
[10] J. Mauer, X. Luo, A. Blanjoie, et al. Reversible methylation of m6Am in the 5 prime cap controls mRNA stability. Nature, 541:371-375, 2017.
[11] B. J. Walters, V. Mercaldo, C. J. Gillon, et al. The role of the RNA demethylase FTO (fat mass and obesity-associated) and mRNA methylation in hippocampal memory formation. Neuropsychopharmacology, 42:1502-1510, 2017.
[12] A. Irisarri, A. Corral, N. Perez-Salvador, et al. FTO inhibition mitigates high-fat diet-induced metabolic disturbances and cognitive decline in SAMP8 mice. Molecular Medicine, 31:73, 2025.
[13] M. S. Ben-Haim, Y. Pinto, S. Moshitch-Moshkovitz, et al. Dynamic regulation of N6,2-prime-O-dimethyladenosine (m6Am) in obesity. Nature Communications, 12:7185, 2021.
[14] A. Spychala and U. Ruther. FTO affects hippocampal function by regulation of BDNF processing. PLoS ONE, 14:e0211937, 2019.
[15] L. Li, L. Zang, F. Zhang, et al. Fat mass and obesity-associated (FTO) protein regulates adult neurogenesis. Human Molecular Genetics, 26:2398-2411, 2017.
[16] R. Chang, S. Zhu, J. Peng, et al. The hippocampal FTO-BDNF-TrkB pathway is required for novel object recognition memory reconsolidation in mice. Translational Psychiatry, 13:349, 2023.
[17] M. Tarullo, G. Fernandez Rodriguez, A. Iaiza, et al. Off-target inhibition of human dihydroorotate dehydrogenase (hDHODH) highlights challenges in the development of FTO inhibitors. ACS Pharmacology and Translational Science, 7(12):4096-4111, 2024.
[18] B. Khatiwada, T. T. Nguyen, J. A. Purslow, and V. Venditti. Solution structure ensemble of human FTO reveals druggable surface pockets at the interface between the N- and C-terminal domain. Journal of Biological Chemistry, 298(5):101907, 2022.
[19] A. Singh, F. Pettini, B. Gianibbi, et al. Structure-based discovery of a non-competitive FTO inhibitor bound to a cryptic site at the domain interface. Journal of Molecular Biology, 438(2):169575, 2026.